Optical mapping has become rather common for applications ranging from genome assembly improvement and validation to structural variation discovery. Validation of the rice genome using Optical map data also helped map centromeres and even span one centromere. However, in some cases centromeres have been found to correspond to regions that have poor mapping of optical maps, presumably due to the presence of tandem repeats that lack unique restriction sites. Without the availability of genetic linkage maps or other evidence, those working with NGS based draft assemblies have speculated that large gaps in the optical map correspond to centromeres or other repeats.
The Human genome has a higher quality as well as availability of various other resources such as genetic linkage maps, BAC's, FISH etc in addition to the availability of optical mapping data. Centromeres have been mapped in the human genome with various other methods and provides an ideal case to investigate the patterns(or lack thereof) shown by optical maps near centromeres. Optical map data for the human and mouse genome from published studies have been made available as bigBed files. This provides a unique resource to understand the behavior of optical maps near centromeres.
First we download the bigBed files for the human genome Hg38 and convert it to bed12 format. This can then be converted to bed format using the convert perl script
wget ftp://ngs.sanger.ac.uk/production/grit/track_hub/hg38/om_align_GM10860.bigBed
wget ftp://ngs.sanger.ac.uk/production/grit/track_hub/hg38/om_align_GM15510.bigBed
wget ftp://ngs.sanger.ac.uk/production/grit/track_hub/hg38/om_align_GM18994.bigBed
for i in om_align_GM10860 om_align_GM15510 om_align_GM18994
do
bigBedToBed "$i".bigBed "$i".bed
perl convertBed.pl "$i".bed > "$i"_full.bed
done
Perl script called covertBed.pl to convert bed12 to bed format.
#!/usr/bin/perl
my $bed12file = $ARGV[0];
open(FILE1, $bed12file);
while($header1=<FILE1>) {
chomp $header1;
my @parts=split(/\t/,$header1);
my @parts2=split(/\,/,$parts[10]);
my @parts3=split(/\,/,$parts[11]);
my $arrSize = @parts2;
for($i=0;$i<$arrSize;$i++){
$start=$parts[1]+$parts3[$i];
$end=$start+$parts2[$i];
print "$parts[0]\t$start\t$end\t$parts2[$i]\n";
}
}
close FILE1;
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -B -A -D hg38 -e 'select chrom,size from chromInfo' > chromhg38.genome
bedtools makewindows -g chromhg38.genome -w 50000|sort -k1,1 -k2,2n > 50kb.wins
cat *_full.bed|sort -k1,1 -k2,2n > full_all.bed
bedtools map -a 50kb.wins -b full_all.bed -c 4 -o mean -null NA > all.mean
bedtools map -a 50kb.wins -b full_all.bed -c 4 -o count -null NA > all.count
Analysis of these numbers across each chromosome suggests that mean fragment size might be a better predictor of centromeres than the count. The position of the centromeres is obtained from the UCSC tables using below command:
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -B -A -D hg38 -e 'select chrom,chromStart,chromEnd from centromeres'|grep -v "chromStart" > centromeres.hg38.genome
for (chr in c("chr1","chr2","chr3","chr4","chr5","chr6","chr7","chr8","chr9","chr10","chr11","chr12","chr13","chr14","chr15","chr16","chr17","chr18","chr19","chr20","chr22","chrX"))
{
read.table(file="all.mean",header=F)->M
read.table(file="centromeres.hg38.genome",header=F)->C
interleave <- function(v1,v2)
{
ord1 <- 2*(1:length(v1))-1
ord2 <- 2*(1:length(v2))
c(v1,v2)[order(c(ord1,ord2))]
}
jpeg(paste(chr,"_OM.jpeg",sep=""))
plot(M$V2[M$V1==chr],M$V4[M$V1==chr],xlab="Position along chromosome",ylab="Mean fragment length in Optical Map",main=chr,col="blue",pch=16)
lines(interleave(C$V2[C$V1==chr],C$V3[C$V1==chr]),rep(0.2,length(interleave(C$V2[C$V1==chr],C$V3[C$V1==chr]))),col="red",lwd=5)
dev.off()
}
















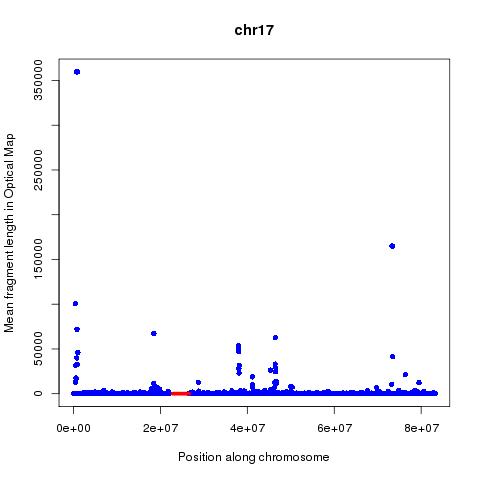





Based on the above analysis can one conclude that it is possible to map Centromeres using Optical mapping data? Far from it, the many false positives and lack of signal in many cases are worrying. The following questions are of importance:
- How does the quality of the genome assembly in the regions adjoining the centromere affect the ability to map centromeres?
- How does the enzyme used and repeat content and base composition of the centromere affect the precision of attempts aimed at mapping the centromere?
No comments:
Post a Comment